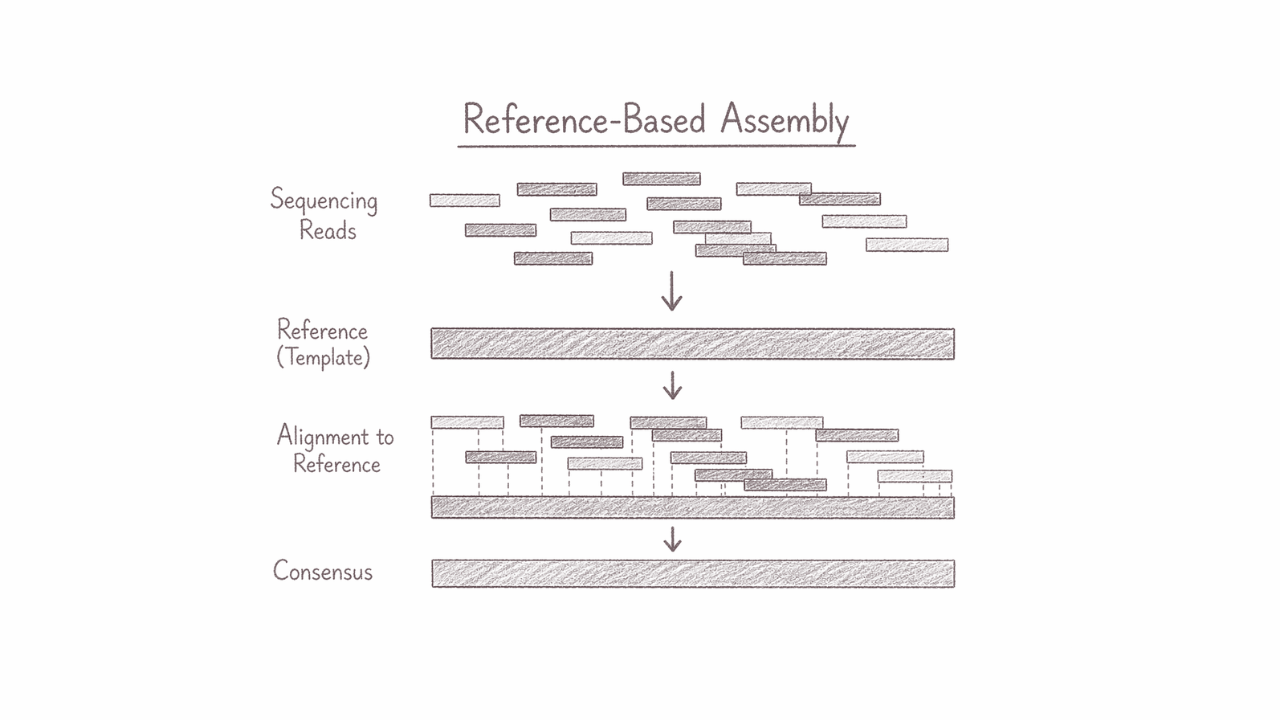
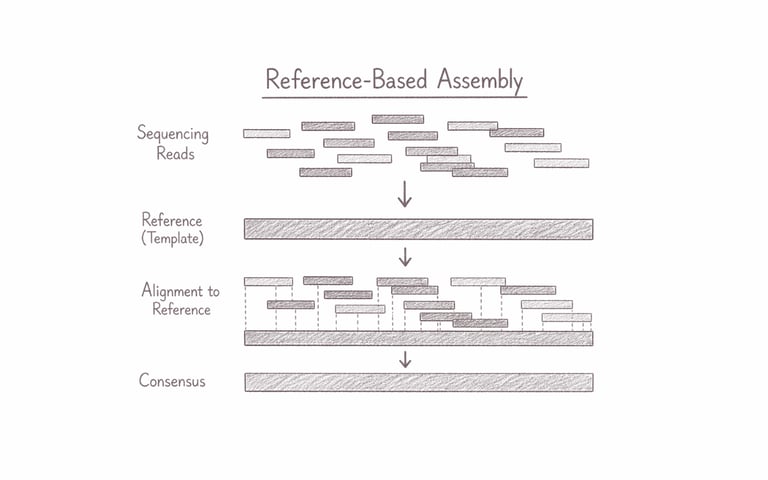
Genome Assembly (reference-based)
Reference-based genome assembly reconstructs a genome by aligning sequencing reads against an existing reference genome from a closely related organism. This approach enables highly efficient and accurate genome reconstruction, making it ideal for identifying genetic variants, comparing strains or populations, and analysing well-characterised species. It is particularly valuable for clinical genomics, evolutionary studies, and large-scale genomic projects where speed, precision, and consistency are essential for generating reliable biological insights.


Procaryote Genome
Rp. 300.000 ($ 20) per sample
7 days


Eucaryote Genome
Start from: Rp. 500.000 ($ 30) per sample
7 days
small-size genome (<150Mbp)

Eucaryote Genome
large-size genome (>150Mbp)
Start from: Rp. 1.000.000 ($ 60) per sample
7 days
MODEL ORGANISM


Eucaryote Genome
large-size genome (>150Mbp)
NON-MODEL ORGANISM
Start from: Rp. 1.500.000 ($ 90) per sample
Start from: 7 days
Default Deliverables
Assembled Genome
FASTA format containing consensus genome sequences reconstructed through alignment against a reference genome, including mapped contigs or polished assemblies.Assembly Metrics Report
Comprehensive quality assessment, including read mapping rate, coverage depth, genome coverage percentage, GC content, consensus quality, and assembly completeness.Annotation Files
GFF3/GTF: gene coordinates and structural features
CDS sequences (FNA): nucleotide coding regions
Protein sequences (FAA): translated gene products
Functional Annotation
Functional characterisation of predicted genes, including Gene Ontology (GO), KEGG pathways, Pfam domains, InterPro annotations, and more.Standard Variant Discovery
Identification of genomic variations, including Average Nucleotide Identity (ANI), aligned portion, single nucleotide polymorphisms (SNPs), insertions and deletions (indels), and structural variants (SVs).Phylogenetic Analysis
Evolutionary relationship analysis based on conserved genes or whole-genome data. Outputs are provided in Newick (.nwk) format and as publication-ready images (.png).
Add-ons
Basecalling
Repeat Annotation
Epigenomic Analysis