Genome Assembly (de novo)
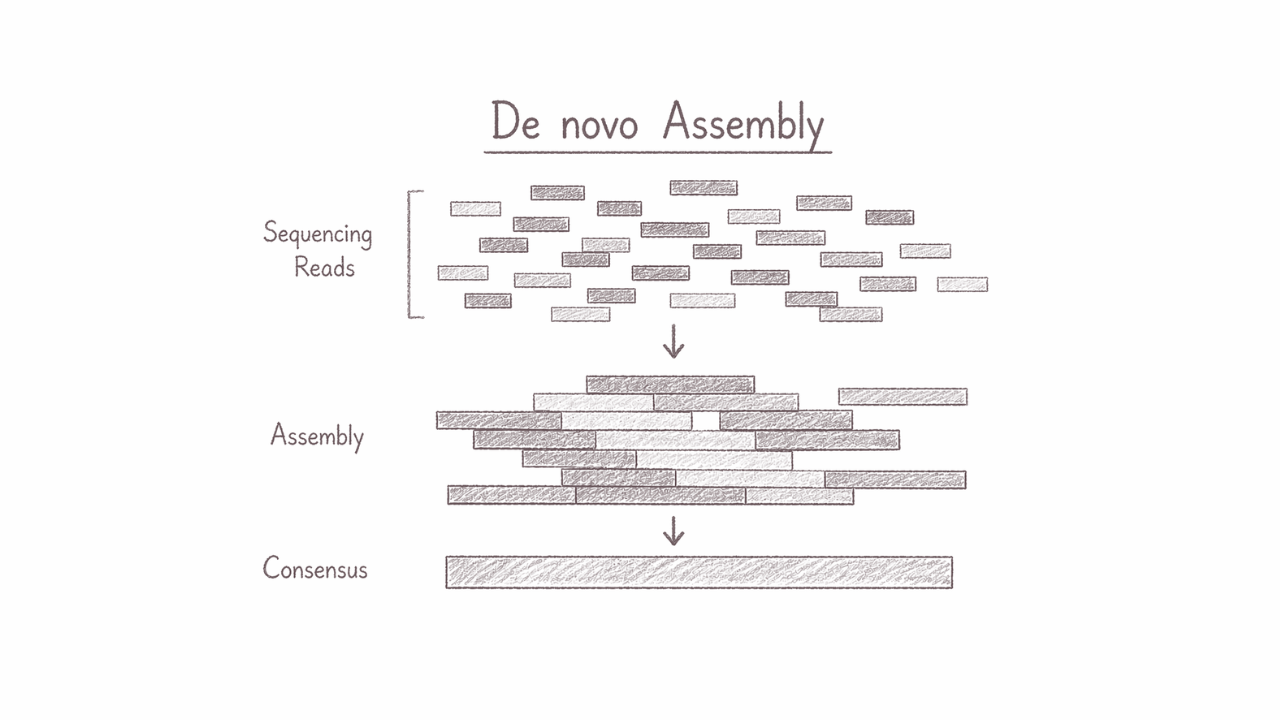
De novo genome assembly reconstructs a complete genome directly from raw sequencing reads, without relying on any reference genome. This approach is essential for exploring previously uncharacterized organisms, uncovering novel genetic architectures, and capturing unique structural variations that reference-based methods may miss. It is particularly powerful for biodiversity studies, microbial discovery, and advancing biotech innovation through truly original genomic insights.
Base Pricing & Turnaround Time


Procaryote Genome
Rp. 1.000.000 ($ 60) per sample
7 days


Eucaryote Genome
Start from: Rp. 1.200.000 ($ 70) per sample
7 days
small-size genome (<150Mbp)


Eucaryote Genome
large-size genome (>150Mbp)
Start from: Rp. 2.000.000 ($ 120) per sample
Start from: 7 days
Default Deliverables
Assembled Genome
FASTA format containing high-quality contigs, scaffolds, or chromosome-level assemblies.Assembly Metrics Report
Comprehensive quality assessment including N50/L50, total genome size, GC content, and genome completeness (BUSCO).Annotation Files
GFF3/GTF: gene coordinates and structural features
CDS sequences (FNA): nucleotide coding regions
Protein sequences (FAA): translated gene products
Functional Annotation
Functional characterisation of predicted genes, including Gene Ontology (GO), KEGG pathways, Pfam domains, InterPro annotations, and more.Standard Variant Discovery
Identification of genomic variations, including Average Nucleotide Identity (ANI), aligned portion, single nucleotide polymorphisms (SNPs), insertions and deletions (indels), and structural variants (SVs).Phylogenetic Analysis
Evolutionary relationship analysis based on conserved genes or whole-genome data. Outputs are provided in Newick (.nwk) format and as publication-ready images (.png).
Add-ons
Basecalling
Hybrid Assembly
Chromosome-level Scaffolding
Repeat Annotation
Epigenomic Analysis