Comparison of AlphaFold 3, Boltz-1, and Chai-1
Comparison of AlphaFold 3, Boltz-1, and Chai-1
Artificial intelligence has rapidly transformed protein structure prediction from one of biology’s hardest problems into a field that now underpins drug discovery and molecular engineering. Traditionally, determining the 3D shapes of proteins and their complexes required years of experimental work with X-ray crystallography or cryo-EM. Today, deep learning models can generate highly accurate predictions in hours, offering insights into how proteins fold, how they interact with ligands or nucleic acids, and how these interactions can be engineered for therapeutic purposes. This revolution began with the breakthrough of AlphaFold, but the landscape has quickly diversified with new models that push the boundaries of speed, openness, and flexibility.
AlphaFold 3 (DeepMind) is the newest version of the protein structure prediction model that has revolutionized structural biology. AlphaFold 2 (2021) opened a new era in protein modeling with high accuracy, but it was still limited to “regular” proteins. To overcome these limits, AlphaFold 3 uses a new diffusion-based architecture that can predict complex biomolecular structures in an integrated way, including proteins, nucleic acids (DNA/RNA), small molecules (ligands), ions, and modified residues. With its “pairformer” architecture, replacing the evoformer of AlphaFold 2, and a diffusion module for atomic coordinates, AlphaFold 3 reaches much higher accuracy in predicting protein–ligand, protein–nucleic acid, and antigen–antibody interactions compared to older specialized methods. Benchmark tests show AlphaFold 3 is superior in almost all categories of biomolecular complexes. This makes AlphaFold 3 very powerful in mapping complex protein structures with high accuracy for drug design and molecular engineering.
Even though it is a breakthrough, AlphaFold 3 also has practical limitations. The model is only available through a non-commercial server and is closed-source, with restrictions on which ligands can be used. Its source code has not been released freely, which limits user access. AlphaFold 3 also requires heavy computation: strong GPUs and long inference times are needed for large complexes. Studies have shown small stereochemical mistakes remain (such as chirality violations of ~4.4%), so expert checking is still needed. Still, the mix of high accuracy and broad coverage makes AlphaFold 3 one of the most influential tools in modern drug discovery, allowing scientists to map drug-target interactions that are very hard to study with traditional physical methods.
Boltz-1 is a collaborative model from MIT’s Jameel Clinic, designed to “democratize” biomolecular interaction modeling. Unlike AlphaFold 3, which is closed, Boltz-1 is fully open-source under an MIT license. Its goal is to create a modeling backbone that anyone can access, while still reaching the same accuracy as AlphaFold 3. Boltz-1 uses a similar diffusion-based deep learning approach but adds innovations in data processing and speed optimization. Evaluations show that Boltz-1 performs at the same level as AlphaFold 3 and Chai-1 on many benchmarks (for example CASP15), producing comparable results in predicting protein–protein and protein–ligand interactions. Optimization techniques, such as local attention, make Boltz-1 faster and more memory-efficient than a standard AlphaFold 3 implementation. Because it is open, Boltz-1 can be directly integrated into academic or industry research without commercial limits, which strongly supports faster drug design and molecular engineering. However, like other generative models, Boltz-1 sometimes produces artifacts (for example, physically impossible overlapping protein chains), especially in very large complexes. The use of relatively small cropping windows (384–512 tokens) also limits the spatial context for very large structures. The Boltz team is working on the next generation, “Boltz-2,” which will also predict ligand binding affinities, opening the door to much faster virtual drug screening.
Chai-1 was developed by Chai Discovery as an open multi-modal foundation model for biomolecular structure prediction. Like Boltz-1, Chai-1 is released for free, including commercial use, under the Apache 2.0 license. Chai-1 supports integrated predictions across proteins, nucleic acids, small ligands, and covalent modifications. A unique feature is that it can run with or without multiple-sequence alignments (MSA). In “single-sequence” mode without MSA, performance is almost the same, which makes it faster in some use cases. Tests show that Chai-1 reached ~77% success in the PoseBusters benchmark (for ligand docking) compared to 76% by AlphaFold 3, and an average LDDT score of 0.849 in CASP15 monomer predictions (compared to 0.801 by ESM3). Chai-1 also shows strong results in multimer predictions: it can model antibody–antigen complexes with accuracy close to AlphaFold-Multimer (DockQ 69.8% vs 67.7%) even without MSA. Moreover, Chai-1 can accept extra experimental input (like binding pocket residues), which improves accuracy by more than ten percentage points in antibody–antigen modeling. Because it is open and cross-platform (available via web interface and code libraries), Chai-1 has been quickly adopted by researchers and companies for drug discovery. It is still new, and much information comes from technical reports, but the team is committed to building more AI foundation models to “predict and reprogram” biochemical interactions. Their long-term mission is to turn biology into an engineering discipline through advanced generative AI.
In terms of structural and interaction prediction performance, all three models are at the top of current technology. AlphaFold 3 sets a new standard with very high accuracy across many complex types, but its access is limited. Boltz-1 and Chai-1 match this accuracy while being open-source, encouraging global collaboration. All three support prediction of protein structures, protein–protein complexes, ligand docking, and nucleic acids with low error rates. Boltz-1 focuses on computational efficiency with its optimizations, while Chai-1 offers more flexibility (no MSA needed) and experimental data integration. In biotechnology, they all have a huge impact: making structure-based drug design and complex molecular engineering more practical. In the future, AlphaFold may expand into dynamic predictions or integration with experimental data (for example, modeling “cellular clocks”), while Boltz-2 will add binding affinity predictions for more precise drug discovery. Chai Discovery also plans to release more foundation models like Chai-2 that are capable of zero-shot de novo antibody design with ~16–20% experimental hit rates and generating binders for half of 52 novel targets from scratch. Together, these tools represent a clear shift in biology into an era of automated design with AI, where the old limits of molecular modelling are being overcome computationally.
As these tools mature, researchers in academia and industry have an unprecedented opportunity to harness them for accelerating discovery in biotechnology. AlphaFold 3, Boltz-1, and Chai-1 already make it possible to explore molecular interactions at scales and speeds unthinkable a decade ago, but there is still vast room for innovation. Scientists can adopt these platforms to design new drugs, model synthetic enzymes, or probe disease mechanisms, and equally, they can contribute to building the next generation of open, accurate, and versatile models. By engaging with this ecosystem, or even developing competing architectures, the community can accelerate biology’s transformation into an engineering discipline where molecular design becomes as programmable and creative as software development itself.
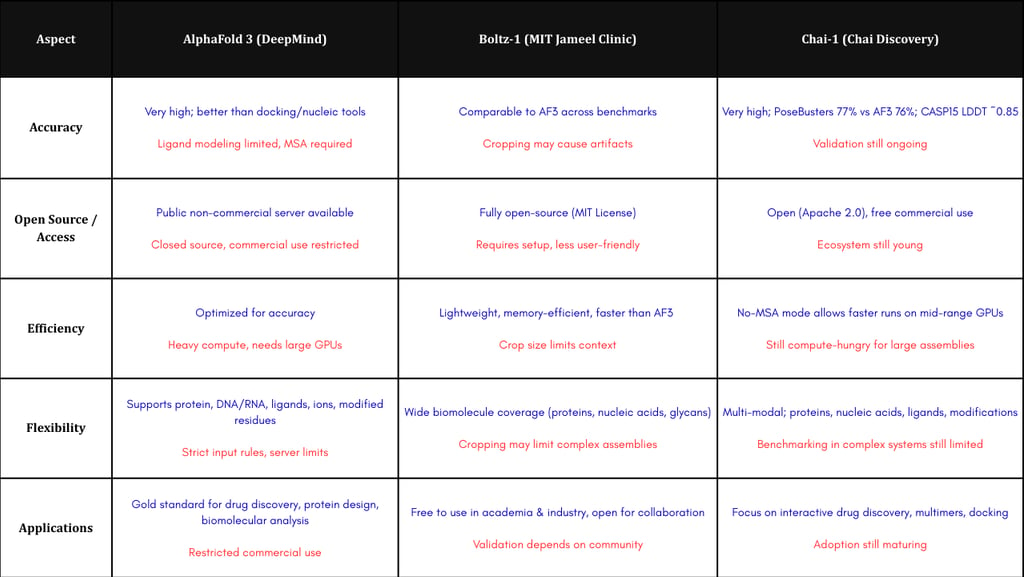
Comparison Table of AlphaFold 3, Boltz-1, and Chai-1

(+62) 815-1304-5845
© 2025. All rights reserved.
Reach us
Got any questions?
Yogyakarta, Indonesia